- Research
- Open Access
- Published:
Parasites & Vectors volume 15, Article number: 493 (2022)
Abstract
Background
The family Capillariidae is a group of thread-like nematodes of 27 genera and over 300 species that infect a great variety of hosts including humans. Among these, some taxa such as the genus Aonchotheca have remained controversial regarding their systematic status for decades. The aim of the current study was to verify Aonchotheca’s systemic status and to further determine whether it is a distinct genus from Capillaria using molecular and phylogenetic analyses.
Results
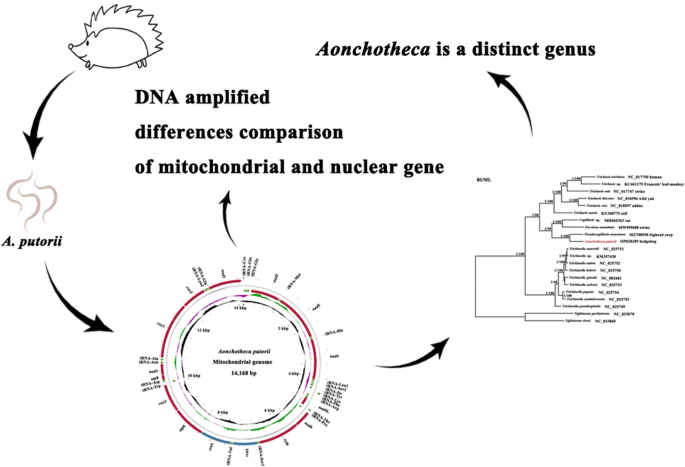
We sequenced the mitochondrial (mt) genome and nuclear small subunit (18S) rRNA gene of Aonchotheca putorii, a representative species of the genus, and investigated its systematic status in Trichinellida using maximum likelihood and Bayesian inference. The differences in amino acid sequences of 13 protein-coding genes were 12.69–67.35% among Aonchotheca, Capillaria, Eucoleus, and Pseudocapillaria with cox1 (12.69%) and atp8 (67.35%) as the most and the least conserved gene, respectively, and the difference of two mt rRNAs was 18.61–34.15%. Phylogenetic analyses of the complete mt genome and 18S rRNAs unequivocally showed that Aonchotheca was a distinct genus from Capillaria.
Conclusions
Large difference exists among Aonchotheca, Capillaria, Eucoleus, and Pseudocapillarias. Aonchotheca putorii is the first species in the genus Aonchotheca for which a complete mitogenome has been sequenced. These data are useful for phylogenetics, systematics and the evolution of Capillariidae.
Graphical Abstract
Background
Capillariidae (Railliet, 1915) is a family of thread-like nematodes traditionally differentiated from others by the characteristic esophagus, male caudal end, and unique morphology of eggs [1,2,3]. So far, over 300 species have been recorded worldwide in almost all groups of vertebrate hosts such as fishes, reptiles, amphibians, birds, mammals, and humans [1, 4, 5]. Capillariids mainly parasitize the internal organs of hosts such as the respiratory and digestive tract, esophagus, stomach, and liver, severely damaging them [1, 3]. However, the identification of Capillariidae to species or even genus by morphology alone is one of the most complicated and challenging tasks among all nematodes. Consequently, controversies on some taxon remain until now [4, 6, 7]. Some taxonomic experts have proposed several new genera and synonymies of capillariids and suggested reclassifying this group [1, 3].
The use of DNA-based molecular techniques is one of the recent advancements for the accurate identification and understanding of nematode evolutionary relationships [8, 9]. The nuclear small subunit of the ribosomal gene (18S rRNA) has been frequently used to analyze phylogenetic relationships among nematodes at different taxonomic levels, especially of some representative capillariids [5, 10,11,12,13,14]. Genome-based phylogenetic analyses such as mitochondrial (mt) genome (mitogenome) are increasingly utilized by the taxonomist for a better understanding of nematodes’ relationships at various taxonomic levels and/or judgment of the discordances and congruences with morphology-based and nuclear rDNA-based phylogenies due to the shortcomings of 18S rRNA [15, 16]. Within the family Capillariidae, the currently available mitogenomes are representatives of the genus Capillaria Zeder, 1800 (MH665363), Eucoleus Dujardin, 1845 (NC_056391), and Pseudocapillaria Freitas, 1959 (MZ708958).
Members of the genus Aonchotheca López-Neyra, 1947, are prevalent in many geographical regions and mainly infect the stomach and intestines of wild carnivores, especially mustelids [17]. They utilize earthworms as a reservoir to enhance their infection to the definitive hosts [18]. The taxonomic status of Aonchotheca putorii (Rudolphi, 1819) López-Neyra, 1947, within Capillariidae has also been revised several times, and it is often considered a species of the genus Capillaria [18]. Intra-specific morphometric variations among capillariids have been frequently observed because of their phenotypic plasticity, physiological and/or immunological effects of different hosts or even processing techniques in different laboratories [19]. Tamaru et al. observed significant nucleotide variation in the 18S rRNA sequence among specimens of A. putorii collected from various wild carnivores in Japan [19]. In contrast, Kołodziej-Sobocinska et al. did not observe significant genetic divergence in it among A. putorii worms collected from either same or different hosts at distant geographical localizations [20]. Hence, the two aims of the current study were to (1) analyze the genetic divergence in the 18S rRNA sequence of A. putorii from different hosts and locations and (2) verify the status of Aonchotheca by comparing mt coding regions and analyzing phylogenetic relationships within the family Capillariidae.
Materials and methods
Parasites and molecular identification
Adult parasites were collected from the small intestine of dead hedgehogs (Erinaceus europaeus Linnaeus, 1758) originated from Beijing, China. The specimens were intensively washed in ultrapure water and physiological saline solution, and then stored in 75% ethanol. They were morphologically identified as capillariid nematodes with the presence of precloacal caudal alae in male worms [5]. For molecular identification, the total genomic DNA was extracted from several worms using QIAamp® DNA Micro Kit (Qiagen, Hilden, Germany) per the manufacturer’s instruction. Polymerase chain reaction (PCR) was applied to amplify mitochondrial (mt) gene cox1 using the primers JB3-JB4.5 as previously described [21] and nuclear 18S rRNA by the newly designed ones (F: 5ʹ-TTG GTG CGT TCG GTT CGC TGT T-3ʹ; R: 5ʹ-CCA AGC GAG CAG CAT CAG TCC A-3ʹ). PCR was carried out in BIO-RAD T100™ thermal cycler (Bio-Rad, Hercules, CA, USA) using the following conditions: 95 ℃ 1 min, followed by 37 cycles of 98 ℃ 10 s, 60 ℃ 30 s and 72 ℃ 1 min, with a final extension at 72 ℃ 8 min. The PCR amplicons were purified using NucleoSpin® Gel and PCR Clean-up (Takara Bio USA, Inc.) and sent to BGI Co., Ltd. (Shenzhen, China) for direct sequencing in both directions. The DNA sequences were assembled by DNAMAN v6.0 (Lynnon Biosoft, USA). These sequences were then used to BLAST search GenBank to confirm their identities and species identification of the worms of DNA origin.
Sequencing and assembling mitochondrial genome
Total genomic DNA was fragmented to about 350 bp. The DNA libraries were sequenced using Illumina Hiseq 6000 platform (Novogene Co. Ltd. Tianjin, China). The raw sequences were in FASTQ format. Clean DNA sequences were obtained by removing adapter sequences, low-quality bases (Phred quality < 5), or uncertain reads with repetitive “N” bases. The partial cox1 sequence derived from PCR amplicons as described earlier was then used as the initial reference to assemble the complete mitogenome sequence of A. putorii using Geneious Prime v.2022.0.1 [22] with the following parameters: (i) minimum overlap within the range of 150–200 bp; (ii) minimum overlap identity of 99%; (iii) maximum gap of 5 bp. The complete circular mitogenome of the roundworm was then verified by PCR with primers as listed (Additional files 1 and 2: Table S1 and Figure S1).
Annotation of mitochondrial genome
The webserver MITOs was used for the preliminary annotation of 37 genes in the above-assembled mitogenome [23]. Initiation/termination codons and gene boundaries for all 13 protein-coding genes (PCGs) were identified by Open Reading Frame (ORF) Finder of NCBI (https://www.ncbi.nlm.nih.gov/orffinder/). Locations and borders of two rRNAs (rrnL and rrnS) were recognized based on Tandem [24] and aligned capillariid sequences. Similarity, sequences, directions, and the secondary structures of 22 transfer RNA genes (tRNAs) were further verified by tRNAscan-SE 2.0 with a cutoff score of 1.0 [25]. MEGA v11.0 was used to verify accuracies of 13 PCGs and infer 13 amino acid sequences, codon usage and relative synonymous codon usage (RSCU) [26]. The values of the non-synonymous (Ka)/synonymous (Ks) substitution ratios were calculated with DnaSP v5 to understand the selective stress in the evolution of the family Capillariidae.
Phylogenetic analyses
The 18 complete mitogenomes in the order Trichinellida available in GenBank were all included in phylogenetic analyses. To better understand the evolution and phylogenetic relationships of Trichinellida lineage, we used Xiphinema pachtaicum Tulaganov, 1938 (GenBank no. NC_033870) and X. rivesi Dalmaso, 1969 (GenBank no. NC_033869) as outgroups [27] (Table 1), as Dorylaimida evolves more rapidly in their mitogenome than Trichinellida within the class Enoplea [10]. The aligned amino acid sequences by MAFFT online server with “L-INS-I” [28] were concatenated into a single alignment database and uploaded to Gbolcks 0.91b [29] to exclude ambiguity and identify more conserved blocks by selecting the option for “more stringent.” Bayesian inference (BI) and maximum likelihood (ML) were applied to phylogenetic analyses. Using Mrbayes 3.2 [30] with default model “JC69,” BI tree was constructed with four independent Markov chains by analyzing 1,000,000 generations and sampling tree every 100 generations. Based on ModelFinder in IQTree v.2.1.3 and ProtTest 3.4.2 [31, 32], “mtZOA + F + R5” and “MtArt + I + G + F” were selected as the most suitable models for ML analysis in IQTree v.2.1.3 and PhyML 3.1, respectively. Additionally, available 18S rRNA sequences within the family Capillariidae were used to analyze nuclear phylogenetic relationships and genetic distances. The 18S rRNA sequences were aligned to a single database and used to constructed phylogenetic analyses based on ML (best model: GTR + I + G) and BI (best model: GTR) with the same steps as above. The alignment database was also applied to generate genetic distances of 18S rRNA using MEGA v11.0 [26].
Results and discussion
First molecular identification of A. putorii in China
The mt cox1 sequence of PCR amplicon of 652 bp (GenBank accession no: OP363931) using primer pair JB3-JB4.5 as previously described [21] showed only 81.7% identities to GenBank no. MH665361 by BLAST search, which was a Capillaria sp. This low identity indicates the parasitic worm may be one member of the family Capillariidae and relate to Capillaria sp. species. We then PCR amplified and sequenced 18S rRNA of 1813 bp. The newly obtained 18S rRNA sequence (GenBank accession no: OP028951) of this nematode showed 100% identity to GenBank no. LC052349 of A. putorii. Therefore, the nematodes recovered from the small intestine of hedgehogs in Beijing, China, are identified as A. putorii, which is the first report in the country to our knowledge. Aonchotheca. putorii has been found in a wide range of geographical locations and several mammalian hosts including minks, weasels, martens, racoons, and even the domestic cats [17].
Divergency of 18S rRNA gene in the family Capillariidae
We then further analyzed the genetic divergence of 18S rRNA of 19 members in the family Capillariidae (Additional file 1: Table S2), which ranged from 0 to 14.1%. The 18S rRNA of Capillaria spinulosa (Linstow, 1890) was the most variable (10.9–14.1%) (Additional file 1: Table S2). Kołodziej-Sobocinska et al. reported no significant genetic divergence of 18S rRNA genes was observed in A. putorii collected from different geographical distributions and various hosts [20]. However, Tamaru et al. found significant nucleotide variation of 18S rRNA genes among A. putorii collected from various animals [19]. Our result showed clear divergence in 18S rRNA sequences of worms recovered from Japan, America, and China. Specifically, comparing currently available 500 bp of 18S rRNA isolated from American A. putorii with those from China and Japan, up to 4.00% divergency was found. Furthermore, up to 8.94% (31 nucleotide substitutions) difference was found between Japanese and Chinese isolates (Additional file 1: Table S3).
First complete mt genome of A. putorii
We then tried to decode the whole mt genome of A. putorii. Due to the lack of mt genome of closely related species, we took an unusual strategy using the newly cox1 DNA sequence just mentioned and next generation sequencing technique as outlined earlier. The complete mitogenome of A. putorii was 14,168 bp in length and had been uploaded into GenBank with the accession number OP028209. It comprised 37 genes (Table 2, Fig. 1) including 13 PCGs (atp6, atp8, cox1-cox3, cytb, nad1-nad6, and nad4L), 2 rRNAs (12S rRNA and 16S rRNA) and 22 tRNAs (Fig. 1). Its genetic arrangement was consistent with those of published Eucoleus annulatus (Molin, 1858) López-Neyra, 1946 (MW999680) [33], and Pseudocapillaria tomentosa (Dujardin, 1843) Lomakin and Trofimenko, 1982 (MZ708958), but different from that of Capillaria sp. (MH665363). The findings indicated at least one inversion between Aonchotheca − Eucoleus − Pseudocapillaria species and Capillaria sp., though four tRNAs (tRNA-Gly, tRNA-Tyr, tRNA-Cys and tRNA-His) were lacking in the Capillaria sp.. The tRNA-Gln was found on the L-strand between tRNA-Cys and tRNA-Gly in A. putorii, E. annulatus, and P. tomentosa, while it was located between nad5 and nad4 in Capillaria sp. (not shown).
The organization of the complete mitochondrial genome of Aonchotheca putorii (Rudolphi, 1819) López-Neyra, 1947. The scale is approximate. Different colors represent different elements; the directions of arrows represent the directions of gene transcriptions; the height of peaks represent the value of GC skew + , GC skew- and GC content. atp6 and atp8 ATP synthase F0 subunits 6 and 8, cytb cytochrome b, cox1‑3 cytochrome c oxidase subunits 1–3, nad1‑6 and nad4L NADH dehydrogenase subunits 1–6 and 4L, rrnS and rrnL small and large subunits of ribosomal RNA
Twenty-five intergenic regions ranging from 1 to 72 bp in length were found scattered throughout the mitogenome. The highest AT content of 84.8% was in intergenic regions, and AT-skew in these regions was positive while the value of GC-skew in them was “0” (the number of G bases was roughly equal to C bases, Table 3). The mitogenomes of nematodes usually contain two non-coding regions (NCRs) with different sizes [15, 33, 34]. The length of AT-rich was 137 bp in Necator americanus Stiles, 1902 [35], 886 bp in Ascaris suum Goeze, 1782 [36], and approximately 7 kb in Hoplolaimus columbus Sher, 1963 [37]. As far as members of the family Capillariidae were concerned, no AT-rich region was found in the mitogenome of A. putorii, but it was 306 bp, 121 bp and 99 bp in E. annulatus [33], Capillaria sp. (MH665363) and P. tomentosa (MZ708958), respectively.
Aonchotheca putorii mitogenome was A-T biased; they accounted for 78.0% (Table 3), which is consistent with other capillariid nematodes [33]. Furthermore, T base (42.2%) was the most frequent and C (9.3%) the least. Both AT-skew and GC-skew included negative and positive values, ranging from − 0.349 (nad6) to 0.031 (NCRs) and from − 0.345 (nad2) to 0.345 (cytb), respectively.
Characteristics of protein-coding genes
Aligning with available sequences of capillariid nematodes (GenBank no. MZ708958, NC_056391 and MH665363), the boundaries of each PCG were determined, and the initiation/termination codons and the directions of translation were identified. For A. putorii, the longest PCG was cox1 (1584 bp), followed by nad5 (1521 bp). The rest were nad4 (1,263 bp), cytb (1136 bp), nad1 (912 bp), atp6 (909 bp), nad2 (900 bp), cox3 (774 bp), cox2 (684 bp), nad6 (459 bp), nad3 (301 bp), nad4L (249 bp) and atp8 (146 bp).
ATN was exclusively used as an initiation codon in all PCGs where N was T, A or G. ATT was the most frequent initiation codon used in six PCGs (nad2, nad6, atp8, nad3, cox1 and nad1), followed by ATA in five (for nad5, nad4, nad4L, atp6 and cox2). The least favored start codon was ATG used only in cytb and cox3. The complete termination codon TAA was the most common one, which was used in all PCGs except cytb, atp8, and nad3, in which incomplete stop codons TA and T were used, respectively. Start codons ATT, ATA and ATG were shared among those species, while TTG was only used in E. annulatus and Capillaria sp., and ATC was unique in P. tomentosa. Complete termination codon TAA was prevalent among those species, but TAG was used in E. annulatus, Capillaria sp. and P. tomentosa excluding A. putorii.
The GC- and AT-skews for mitogenome are calculated as a measure of the compositional asymmetry and effective assistant of replication [38, 39]. For the A. putorii mitogenome, the AT- and GC-skews were − 0.082 and − 0.157, respectively, which showed no clear trend of A or T bases, but a significant use of C bases compared with other nematodes [40]. Similar bases trend also showed in PCGs. AT and GC-skews of 13 PCGs were from − 0.349 (nad6) to 0.071 (nad4L) and from − 0.345 (nad2) to 0.345 (cytb) (Table 3), indicating a trend of A bases among those PCGs that would have an impact on RSCU. RSCU represents an intuitive reflection of the use of codon bias, and a higher RSCU indicates a higher codon usage bias. Within codons encoding amino acids, there was an obvious bias of T- or A-rich codons. For example, Leu was encoded by TTA, TTG, CTT, CTC, CTA and CTG, but the RSCU value of TTA was 3.91 higher than that with C and G residues (Table 4).
In PCGs, Ka/Ks test was helpful for better understanding the selective constraints, evolutionary changes, sexual selection and disease resistance [41]. Generally, the ratio of Ka/Ks being > 1 represents rapid evolution of a protein gene and a positive selection and changes in protein’s functions. In contrast, < 1 indicates a negative selection and selectively purified among genes [42, 43]. In the present study, the Ka/Ks ratios of the genes atp6, cox3, nad3, nad4 and nad6 ranged from 1.07 to 1.72 (Fig. 2), indicating these genes had experienced a positive selection or had undergone functional changes. The Ka/Ks values of atp8, cox1, cox2, cytb, nad1, nad2, nad4L and nad5 were < 1 (Fig. 2), suggesting they were highly constrained within the family Capillariidae. Consistent with previous reports, the findings also indicated that cox1 was selectively purified and might play an adaptive role in the evolutionary process of Capillariidae species, and it can be used as useful marker to identify and distinguish capillariid species. Interestingly, atp8 gene was not only found in those species, but under purifying selection across Capillariidae, though it was variable protein-gene or even missing in some nematodes [44].
Substitution ratios in the mitochondrial genomes of capillariid nematodes. The rate of non-synonymous (Ka), the rate of synonymous (Ks) substitutions and the respective ratios (Ka/Ks) for individual protein-coding genes are shown
Transfer and ribosomal RNA genes
All 22 tRNAs were obtained in the present study ranging from 54 to 71 bp in length. The predicted secondary structures of most tRNAs identified in this study were similar to those found in other nematodes, and the TΨC stem loop and variable loop were replaced by a TV-replacement loop structure, which was common in the mitogenomes of nematodes excluding Trichinella spiralis (Owen, 1835) Railliet, 1895 [44,45,46]. Consistent with previous reports, the predicted structure of tRNA-Ser (AGN and UCN) lacked the DHU-arm, which was replaced by 4–5 nucleotide residues [47, 48]. In the present study, 9 of 20 tRNAs (tRNA-His, tRNA-Ile, tRNA-Arg, tRNA-Lys, tRNA-LeuCUN, tRNA-LeuUUR, tRNA-Met, tRNA-Trp, and tRNA-Tyr) presented relatively standard “cloverleaf” secondary structures like those of T. spiralis [44] (Fig. 3). The length of nine TΨC stems was from 2 to 5 bp, and the length of variable arms between the anticodon loop and TΨC stem-loop ranged from 2 to 8 bp.
The secondary structures of 22 tRNA of Aonchotheca putorii mitochondrial genome. Eleven tRNAs have a typical TV-loop and nine tRNAs relatively standard “cloverleaf” structures; Leu1 and Leu2 for codon families CUN and UUR, respectively; S1 and S2 for codon families UCN and AGN, respectively
The locations and boundaries of rrnL (16S rRNA) and rrnS (12S rRNA) were identified by alignment with available Capillariidae species (NC_056391). The typical arrangement of the rrnL was located between tRNA-Val and atp6, and the rrnS was situated in tRNA-SerAGN and tRNA-Val, similar to Capillaria sp. (MH665363), E. annulatus (MW999680) [33] and P. tomentosa (MZ708958). The length of these two rRNAs was 864 bp and 693 bp, respectively (Table 2).
Comparative analyses of mitochondrial sequence and nuclear 18S rRNA
Comparisons of nucleotide sequences of mt genes are listed in Table 5, as were amino acids of PCG. Transfer RNA genes were not included in the table because Capillaria sp. missed four of them (tRNA-Tyr, tRNA-His, tRNA-Gly and tRNA-Cys). Obvious differences at both nucleotide and amino acid sequences were observed in all 13 PCG. Specifically, the differences of nucleotide and amino acid between A. putorii and Capillaria sp., A. putorii and E. annulatus, and A. putorii and P. tomentosa were 21.96–44.37%, 23.39–48.23% and 17.57–38.36% (Table 5) and 17.80–66.00%, 19.89–67.35% and 12.69–58.33%, respectively (Table 5). Furthermore, cox1 and atp8 were the most and the least conserved PCG, respectively. The differences of rRNAs nucleotide sequences were 18.61–30.99% in rrnL and 22.03–34.15% in rrnS (Table 5). It is acceptable that divergence of mt DNA sequences between species in nematodes was 10–20% [49]. The differences observed in the current study in nucleotide sequences of all 13 PCG and two rRNA genes ranged from 17.57 to 48.23%, and the divergences in amino acids were from 12.69 to 67.35% clearly indicated A. putorii was a distinct species from other Capillariidae species. The differences among Aonchotheca, Capillaria, Eucoleus, and Pseudocapillaria were higher than species level and further suggested Aonchotheca was a distinct genus.
Phylogenetic analyses
BI and ML trees of 18S rRNAs of Capillariidae species (Additional file 2: Figure S1) showed similar topologies to previous studies [5, 50]. These indicated some Capillaria species did not belong to this genus. The tree further proved that genera Capillaria and Eucoleus were all monophyly and more related, the genus Aonchotheca might be paraphyly, and results also showed a closer relationship among genera Aonchotheca, Calodium, Pearsonema, Pseudocapillaria, and Baruscapillaria with moderate supports (Additional file 3: Figure S2).
Using X. pachtaicum and X. rivesi as the outgroups in analyzing phylogenetic relationships within class Trichinellida, both BI and ML trees displayed similar topological and systematic relationships (Fig. 4), which was consistent with previous data [33, 51]. The families Capillariidae, Trichinellidae and Trichuridae were clearly in their own clades. In accord with previous studies based on complete mitogenome or 18S rRNA sequences [33, 50, 51], our results also showed those families were monophyletic with Capillariidae and Trichuridae forming sister taxa of high statistical support (Bpp = 1, Bf ≥ 98).
Phylogenetic relationships among 18 species of Trichinellida nematodes inferred from Bayesian and maximum likelihood analyses of deduced amino acid sequences of 12 mitochondrial proteins. Xiphinema pachtaicum (GenBank no. NC_033870) and Xiphinema rivesi (GenBank no. NC_033869) were used as outgroups. Bayesian posterior probability (Bpp) and bootstrap frequency (Bf) values were indicated at nodes. The former of the phylogenetic tree represents parasitic nematode, and the latter represents their hosts. Most Trichinellidae strain species were maintained by serial passage in female CD1 mice, and they were not labeled in the figure
In the family Capillariidae, Capillaria was a sister taxon genus to Eucoleus, Pseudocapillaria and Aonchotheca, of high support (Bpp = 1, Bf ≥ 99). Furthermore, there was a closer relationship between genera Aonchotheca and Pseudocapillaria, and between Capillaria and Eucoleus, which was similar to the previous results by analyzing cox1 and 18S rRNA [4, 5, 50, 51], indicating cox1 and 18S rRNA genes were useful markers to mark capillariids. In addition, the distances between the Capillaria species (Capillaria sp.) and the other three capillariid nematodes (E. annulates, P. tomentosa and A. putorii) were longer than the distances within other trichinellid species, which further clarified long genetic distance among those genera. These phylogenetic results also indicated that Eucoleus, Pseudocapillaria, and Aonchotheca were three distinct genera from Capillaria, supporting the accuracy of Moravec taxonomic revision [3, 5, 50]. However, there are still taxonomic controversies within Capillariidae concerning phylogenomics and systematic status [4, 19, 33] due to the limited numbers of complete mitogenome in other Capillariidae species, like Baruscapillaria, Calodium, Paracapillaria and Pearsonema.
Conclusion
We have revealed details of the mitogenome of A. putorii and showed that Aonchotheca is a distinct genus from Capillaria. Our data indicate mitogenome is an ideal tool for analyzing the phylogenetic relationships and dealing with systematic controversy. In addition, nuclear DNA of 18S rRNA agrees with the phylogenetic results of mitogenome. Though the molecular information in capillariids has steadily increased recently, mt DNA sequences are still in great demand to reveal evolutionary rate, phylogenomic and systematic relationships within this family.
Availability of data and materials
The complete mitochondrial genome and 18S rRNA sequences of Aonchotheca putorii from the hedgehogs have been deposited in the GenBank under the accession numbers OP028951 and OP028209, respectively.
References:
-
Gibbons LM. Keys to the nematode parasites of vertebrates. Oxon: CAB International; 2010.
-
Hodda M. Animal biodiversity: an outline of higher-level classification and survey of taxonomic richness. Zootaxa. 2011;3148:63–95.
-
Moravec F. Proposal of a new systematic arrangement of nematodes of the family Capillariidae. Folia Parasitol. 1982;29:119–32.
-
Borba VH, Machado-Silva JR, Le Bailly M, Iñiguez AM. Worldwide paleodistribution of Capillariid parasites: paleoparasitology, current status of phylogeny and taxonomic perspectives. PLoS ONE. 2019;14:e0216150.
-
Sakaguchi S, Yunus M, Sugi S, Sato H. Integrated taxonomic approaches to seven species of Capillariid nematodes (Nematoda: Trichocephalida: Trichinelloidea) in poultry from Japan and Indonesia, with special reference to their 18S rDNA phylogenetic relationships. Parasitol Res. 2020;119:957–72.
-
de Carvalho EL, Santana RLS, da Pinheiro RHS, Giese EG. Eucoleus contortus (Nematoda: Capillariidae), a parasite of Cairina moschata domestica (Anseriformes: Anatidae) on Marajó Island, Pará State, in Brazilian Amazon. Rev Bras Parasitol Vet. 2019;28:692–9.
-
Moravec F, Justine JL. Paracapillaria (Paracapillaria) gastrica n. sp. (Nematoda: Capillariidae) from the marine fish Synodus variegatus Lacépède (Synodontidae, Aulopiformes) off New Caledonia. Syst Parasitol. 2020;97:157–63.
-
Ahmed N, Tonelli L, Labagnara G, Doglioni C, Pedica F. Subcutaneous dirofilariosis in Italy: a diagnostic hypothesis to remind when the anamnesis is misleading. Pathologica. 2022;114:174–7.
-
Nisa RU, Tantray AY, Shah AA. Shift from morphological to recent advanced molecular approaches for the identification of nematodes. Genomics. 2022;114:110295.
-
Blaxter ML, De Ley P, Garey JR, Liu LX, Scheldeman P, Vierstraete A, et al. A molecular evolutionary framework for the phylum Nematoda. Nature. 1998;392:71–5.
-
Meldal BHM, Debenham NJ, De Ley P, De Ley IT, Vanfleteren JR, Vierstraete AR, et al. An improved molecular phylogeny of the Nematoda with special emphasis on marine taxa. Mol Phylogenet Evol. 2007;42:622–36.
-
Blaxter M, Koutsovoulos G. The evolution of parasitism in Nematoda. Parasitology. 2015;142:S26-39.
-
Holterman M, Schratzberger M, Helder J. Nematodes as evolutionary commuters between marine, freshwater and terrestrial habitats. Biol J Linn Soc. 2019;128:756–67.
-
Svitin R, Bullard S, Dutton H, Netherlands E, Syrota Y, Verneau O, et al. Pseudocapillaria (Ichthyocapillaria) bumpi n. sp. (Nematoda: Capillariidae) parasitising West African lungfish Protopterus annectens (Owen, 1839) (Lepidosireniformes: Protopteridae) in Mozambique and its phylogenetic position within Capillariid nematodes. Acta Parasitol. 2021;66:1204–11.
-
Kim T, Lee Y, Kil HJ, Park JK. The mitochondrial genome of Acrobeloides varius (Cephalobomorpha) confirms non-monophyly of Tylenchina (Nematoda). PeerJ. 2020;8:e9108.
-
Ahmed M, Holovachov O. Twenty years after De Ley and blaxter-how far did we progress in understanding the phylogeny of the phylum Nematoda? Anim Open Access J MDPI. 2021;11:3479.
-
Zalewski A, Kołodziej-Sobocińska M, Bartoń KA. A tale of two nematodes: climate mediates mustelid infection by nematodes across the geographical range. Int J Parasitol Parasites Wildl. 2022;17:218–24.
-
Butterworth EW, Beverley-Burton M. The taxonomy of Capillaria spp. (Nematoda: Trichuroidea) in carnivorous mammals from Ontario, Canada. Syst Parasitol. 1980;1:211–36.
-
Tamaru M, Yamaki S, Jimenez LA, Sato H. Morphological and molecular genetic characterization of three Capillaria spp. (Capillaria anatis, Capillaria pudendotecta, and Capillaria madseni) and Baruscapillaria obsignata (Nematoda: Trichuridae: Capillariinae) in avians. Parasitol Res. 2015;114:4011–22.
-
Kołodziej-Sobocińska M, Tokarska M, Zalewska H, Popiołek M, Zalewski A. Digestive tract nematode infections in non-native invasive American mink with the first molecular identification of Molineus patens. Int J Parasitol Parasites Wildl. 2021;14:48–52.
-
Zhu X, Spratt DM, Beveridge I, Haycock P, Gasser RB. Mitochondrial DNA polymorphism within and among species of Capillaria sensu lato from Australian marsupials and rodents. Int J Parasitol. 2000;30:933–8.
-
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinforma Oxf Engl. 2012;28:1647–9.
-
Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69:313–9.
-
Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80.
-
Chan PP, Lin BY, Mak AJ, Lowe TM. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021;49:9077–96.
-
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9.
-
Palomares-Rius JE, Cantalapiedra-Navarrete C, Archidona-Yuste A, Blok VC, Castillo P. Mitochondrial genome diversity in dagger and needle nematodes (Nematoda: Longidoridae). Sci Rep. 2017;7:41813.
-
Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019;20:1160–6.
-
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–52.
-
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
-
Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinforma Oxf Engl. 2011;27:1164–5.
-
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9.
-
Deng Y, Suleman, Zhang Y, Nie Y, Fu Y, Liu G. The complete mitochondrial genome of Capillariid nematodes (Eucoleus annulatus): a novel gene arrangement and phylogenetic implications. Vet Parasitol. 2021;296:109476.
-
Jex AR, Hall RS, Littlewood DTJ, Gasser RB. An integrated pipeline for next-generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Res. 2010;38:522–33.
-
Hu M, Chilton NB, Gasser RB. The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea). Int J Parasitol. 2002;32:145–58.
-
Okimoto R, Macfarlane JL, Clary DO, Wolstenholme DR. The mitochondrial genomes of two nematodes, Caenorhabditis elegans and Ascaris suum. Genetics. 1992;130:471–98.
-
Ma X, Agudelo P, Richards VP, Baeza JA. The complete mitochondrial genome of the Columbia lance nematode, Hoplolaimus columbus, a major agricultural pathogen in North America. Parasit Vectors. 2020;13:321.
-
Reyes A, Gissi C, Pesole G, Saccone C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol Biol Evol. 1998;15:957–66.
-
Sahyoun AH, Bernt M, Stadler PF, Tout K. GC skew and mitochondrial origins of replication. Mitochondrion. 2014;17:56–66.
-
Xie Y, Zhang Z, Niu L, Wang Q, Wang C, Lan J, et al. The mitochondrial genome of Baylisascaris procyonis. PLoS ONE. 2011;6:e27066.
-
Fay JC, Wu CI. Sequence divergence, functional constraint, and selection in protein evolution. Annu Rev Genomics Hum Genet. 2003;4:213–35.
-
Li Z, Liu Z, Wei Y, Liu Y, Xing L, Liu M, et al. Genome-wide identification of the MIOX gene family and their expression profile in cotton development and response to abiotic stress. PLoS ONE. 2021;16:e0254111.
-
Meganathan PR, Dubey B, Batzer MA, Ray DA, Haque I. Complete mitochondrial genome sequences of three Crocodylus species and their comparison within the order Crocodylia. Gene. 2011;478:35–41.
-
Lavrov DV, Brown WM. Trichinella spiralis mtDNA: a nematode mitochondrial genome that encodes a putative ATP8 and normally structured tRNAs and has a gene arrangement relatable to those of coelomate metazoans. Genetics. 2001;157:621–37.
-
He Y, Jones J, Armstrong M, Lamberti F, Moens M. The mitochondrial genome of Xiphinema americanum sensu stricto (Nematoda: Enoplea): considerable economization in the length and structural features of encoded genes. J Mol Evol. 2005;61:819–33.
-
Wolstenholme DR, Okimoto R, Macfarlane JL. Nucleotide correlations that suggest tertiary interactions in the TV-replacement loop-containing mitochondrial tRNAs of the nematodes, Caenorhabditis elegans and Ascaris suum. Nucleic Acids Res. 1994;22:4300–6.
-
Hu M, Chilton NB, Gasser RB. The mitochondrial genome of Strongyloides stercoralis (Nematoda) idiosyncratic gene order and evolutionary implications. Int J Parasitol. 2003;33:1393–408.
-
Liu GH, Wu CY, Song HQ, Wei SJ, Xu MJ, Lin RQ, et al. Comparative analyses of the complete mitochondrial genomes of Ascaris lumbricoides and Ascaris suum from humans and pigs. Gene. 2012;492:110–6.
-
Blouin MS. Molecular prospecting for cryptic species of nematodes: mitochondrial DNA versus internal transcribed spacer. Int J Parasitol. 2002;32:527–31.
-
Guardone L, Deplazes P, Macchioni F, Magi M, Mathis A. Ribosomal and mitochondrial DNA analysis of Trichuridae nematodes of carnivores and small mammals. Vet Parasitol. 2013;197:364–9.
-
Feldman SH, Ramirez MP. Molecular phylogeny of Pseudocapillaroides xenopi (Moravec et Cosgrov 1982) and development of a quantitative PCR assay for its detection in aquarium sediment. J Am Assoc Lab Anim Sci. 2014;53:668–74.
Funding
This study was supported by the Training Program for Excellent Young Innovators of Changsha (grant no. KQ2106044).
Author information
Authors and Affiliations
Contributions
CY, GHL, YPD and S conceived and designed the study and critically revised the manuscript. YPD and RL performed the experiments. YPD, CY and GHL analyzed the data. YPD drafted the manuscript. XLZ, LYL and YTF helped in study design, study implementation and manuscript preparation. All authors read and approved the final manuscript.
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The designed primers were used to amplify the complete mitochondrial sequence of Aonchotheca putorii. Table S2 The uncorrected paired genetic distance of 18S rRNAs among the Capillariidae species. Table S3 The uncorrected paired genetic distance of 18S rRNAs among the Aonchotheca putorii from different regions.
The electrophoretogram of the verified complete mitochondrial genome of Aonchotheca putorii. M: 5000 marker; -: negative control; Lane 1: tRNA-Gly – nad4; Lane 2: nad4 – nad4L; Lane 3: rrnS – atp8; Lane 4: atp8 – nad1; Lane 5: nad4L – rrnS.
The phylogenetic analyses based on 18S rRNA among the family Capillariidae using Bayesian posterior probability (Bpp) and Bootstrap frequency (Bf) values were indicated at nodes. The former of the phylogenetic tree represents parasitic nematodes, and the latter represents their hosts.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.